Computational Genomics with R
Check out our new book on Computational Genomics and learn how to analyze your data with R

Check out our new book on Computational Genomics and learn how to analyze your data with R
)
I’m a bioinformatics scientist and the head of Bioinformatics and Omics Data Science Platform at the Berlin Institute of Medical Systems Biology, Max Delbrück Center in Berlin. I have been developing computational methods for analyzing and integrating large-scale genomics data sets since 2002. I mainly use machine learning and statistics to uncover patterns related to important biological variables such as disease state and type. I spent some time in the USA, Norway, Turkey, Japan, and Switzerland in order to pursue research work and education related to statistics, machine learning and bioinformatics.
The underlying aim of my current work is utilizing complex molecular signatures to provide decision support systems for disease diagnostics and biomarker discovery. In addition to the research efforts and managing a scientific lab, since 2015, I have been organizing and teaching at computational genomics courses in Berlin with participants from across the world.
PhD in Genomics & Bioinformatics, 2010
University of Bergen, Norway
BSc in Bioengineering (Bioinformatics track), 2005
Sabanci University, Istanbul, Turkey
Some of the projects I have contributed as a lead or main driver
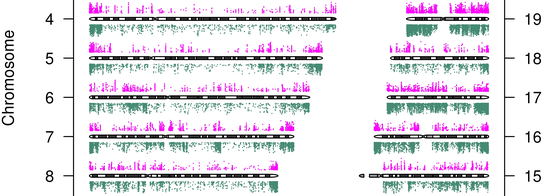
Showed specific AML patients have genome-wide defects in their DNA methylation profiles, this could be exploited for …
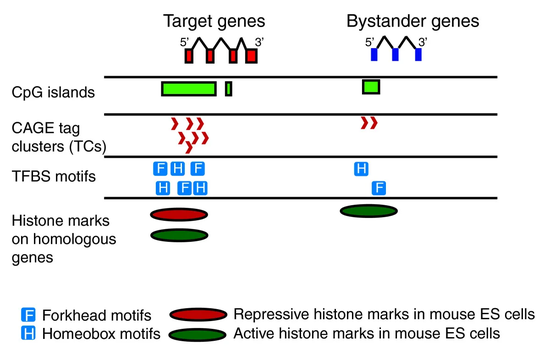
We have identified promoter features of genes under long range gene regulation using statistics and bioinformatics methods.
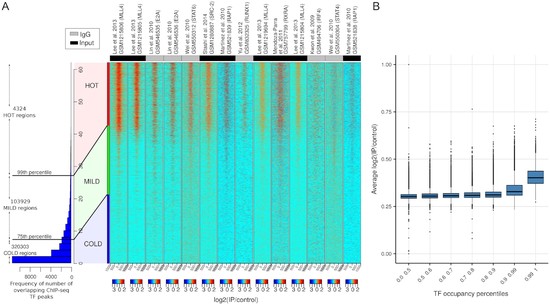
We showed high-occupancy target regions associated with ChIP-seq noise using ML and stats based methods.

Deep learning method for integrating multi-omics data from cancer genomics. Applied on colorectal cancer for refining subtypes.
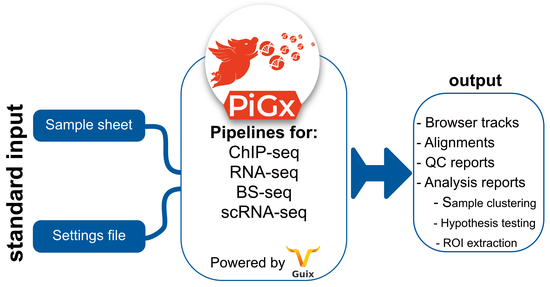
Reproducible, scalable and secure pipelines for genomics | RNA-seq, scRNA-seq, ChIP-seq, BS-seq

Integrate genomic data sets via meta-region plots and heatmaps. Works with BAM, BigWig, Bed files

Analyze methylation data from high-throughput BS-seq experiments
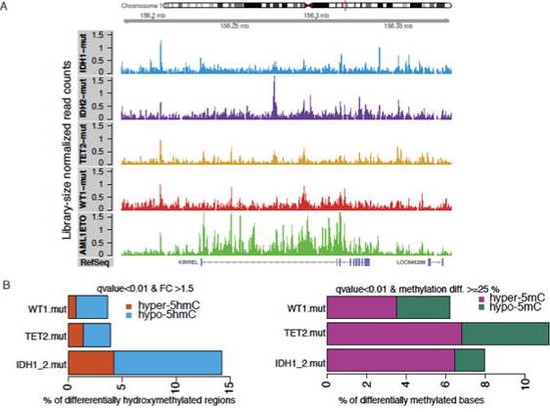
We showed WT1 mutation causes similar epigenome defects to IDH1/2 and TET2 mutations in leukemia
Preprints and peer-reviewed publications
Showed specific AML patients have genome-wide defects in their DNA methylation profiles, this could be exploited for …
We have identified promoter features of genes under long range gene regulation using statistics and bioinformatics methods.
We showed high-occupancy target regions associated with ChIP-seq noise using ML and stats based methods.
Deep learning method for integrating multi-omics data from cancer genomics. Applied on colorectal cancer for refining subtypes.
Reproducible, scalable and secure pipelines for genomics | RNA-seq, scRNA-seq, ChIP-seq, BS-seq
Integrate genomic data sets via meta-region plots and heatmaps. Works with BAM, BigWig, Bed files
Analyze methylation data from high-throughput BS-seq experiments
We showed WT1 mutation causes similar epigenome defects to IDH1/2 and TET2 mutations in leukemia

Posts on different platforms
Showed specific AML patients have genome-wide defects in their DNA methylation profiles, this could be exploited for …
We have identified promoter features of genes under long range gene regulation using statistics and bioinformatics methods.
We showed high-occupancy target regions associated with ChIP-seq noise using ML and stats based methods.
Deep learning method for integrating multi-omics data from cancer genomics. Applied on colorectal cancer for refining subtypes.
Reproducible, scalable and secure pipelines for genomics | RNA-seq, scRNA-seq, ChIP-seq, BS-seq
Integrate genomic data sets via meta-region plots and heatmaps. Works with BAM, BigWig, Bed files
Analyze methylation data from high-throughput BS-seq experiments
We showed WT1 mutation causes similar epigenome defects to IDH1/2 and TET2 mutations in leukemia